Human African Trypanosomiasis (HAT), caused by Trypanosoma brucei gambiense or T. brucei rhodesiense, and malaria, caused by Plasmodium species, are major protozoan parasitic diseases in developing countries, where they are a leading cause of mortality. Conventional treatments are inadequate mainly due to toxicity issues and to the emergence of resistance, so there is a desperate need for new and better drugs.
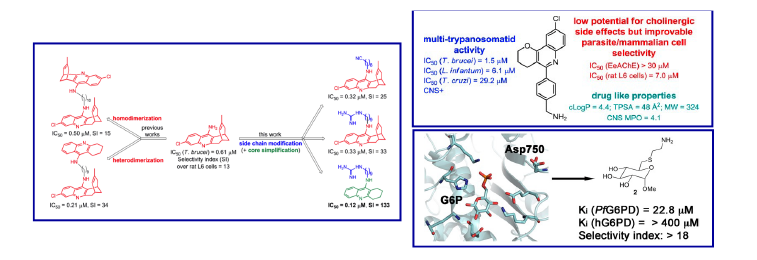
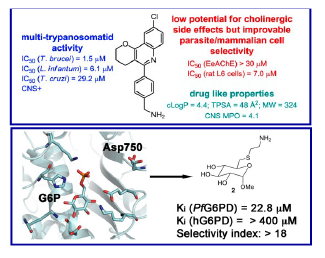
Our work in this field focuses on the synthesis of several classes of antitrypanosomal aminoquinoline-based homo- and hetero-dimeric compounds, multicomponent reaction-based synthesis of quinoline-based derivatives with multi-trypanosomatid activity, and structure-based design of novel antimalarial glucose-6-phosphate dehydrogenase inhibitors. We are also exploring the use of protein aggregation inhibitors as innovative drug candidates against malaria and leishmaniasis.